Pathophysiology
Genetics
Cancers are caused by a series of mutations. Each mutation alters the behavior of the cell somewhat. Cancer is fundamentally a disease of tissue growth regulation. For a normal cell to transform into a cancer cell, the genes that regulate cell growth and differentiation must be altered.
The affected genes are divided into two broad categories. Oncogenes are genes that
promote cell growth and reproduction. Tumor suppressor genes are genes that inhibit cell division and survival. Malignant transformation can occur through the formation of novel oncogenes, the inappropriate over-expression of normal oncogenes, or by the under-expression or disabling of tumor suppressor genes. Typically, changes in multiple genes are required to transform a normal cell into a cancer cell.
Genetic changes can occur at different levels and by different mechanisms. The gain or loss of an entire chromosome can occur through errors in mitosis. More common are mutations, which are changes in the nucleotide sequence of genomic DNA.
Large-scale mutations involve the deletion or gain of a portion of a chromosome. Genomic amplification occurs when a cell gains copies (often 20 or more) of a small chromosomal locus, usually containing one or more oncogenes and adjacent genetic material. Translocation occurs when two separate chromosomal regions become abnormally fused, often at a characteristic location. A well-known example of this is the Philadelphia chromosome, or translocation of chromosomes 9 and 22, which occurs in chronic myelogenous leukemia and results in production of the BCR-abl fusion protein, an oncogenic tyrosine kinase.
Small-scale mutations include point mutations, deletions, and insertions, which may occur in the promoter region of a gene and affect its expression, or may occur in the gene’s coding sequence and alter the function or stability of its protein product. Disruption of a single gene may also result from integration of genomic material from a DNA virus or retrovirus, leading to the expression of viral oncogenes in the affected cell and its descendants.
Replication of the data contained within the DNA of living cells will probabilistically result in some errors (mutations). Complex error correction and prevention are built into the process and safeguard the cell against cancer. If a significant error occurs, the damaged cell can self-destruct through programmed cell death, termed apoptosis. If the error control processes fail, then the mutations will survive and be passed along to daughter cells.
Some environments make errors more likely to arise and propagate. Such environments can include the presence of disruptive substances called carcinogens, repeated physical injury, heat, ionising radiation, or hypoxia.
The errors that cause cancer are self-amplifying and compounding, for example:
- A mutation in the error-correcting machinery of a cell might cause that cell and its children to accumulate errors more rapidly.
- A further mutation in an oncogene might cause the cell to reproduce more rapidly and more frequently than its normal counterparts.
- A further mutation may cause loss of a tumor suppressor gene, disrupting the apoptosis signaling pathway and immortalizing the cell.
- A further mutation in the signaling machinery of the cell might send error-causing signals to nearby cells.
The transformation of a normal cell into cancer is akin to a chain reaction caused by initial errors, which compound into more severe errors, each progressively allowing the cell to escape more controls that limit normal tissue growth. This rebellion-like scenario is an undesirable survival of the fittest, where the driving forces of evolution work against the body’s design and enforcement of order. Once cancer has begun to develop, this ongoing process, termed clonal evolution, drives progression towards more invasive stages. Clonal evolution leads to intra-tumour heterogeneity (cancer cells with
heterogeneous mutations) that complicates designing effective treatment strategies and requires an evolutionary approach to designing treatment.
Characteristic abilities developed by cancers are divided into categories, specifically evasion of apoptosis, self-sufficiency in growth signals, insensitivity to anti-growth signals, sustained angiogenesis, limitless replicative potential, metastasis, reprogramming of energy metabolism and evasion of immune destruction.
Epigenetics
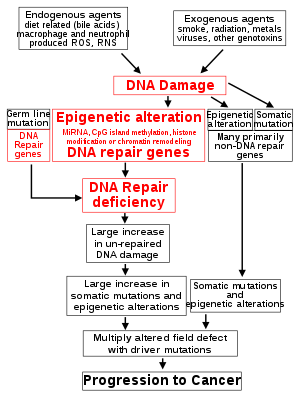
The central role of DNA damage and epigenetic defects in DNA repair genes in carcinogenesis The classical view of cancer is a set of diseases driven by progressive genetic abnormalities that include mutations in tumor-suppressor genes and oncogenes, and in chromosomal abnormalities. A role for epigenetic alterations was identified in the early 21st century.
Epigenetic alterations are functionally relevant modifications to the genome that do not change the nucleotide sequence. Examples of such modifications are changes in DNA methylation (hypermethylation and hypomethylation), histone modification and changes in chromosomal architecture (caused by inappropriate expression of proteins such as HMGA2 or HMGA1). Each of these alterations regulates gene expression without altering the underlying DNA sequence. These changes may remain through cell divisions, endure for multiple generations, and can be considered as equivalent to mutations.
Epigenetic alterations occur frequently in cancers. As an example, one study listed protein coding genes that were frequently altered in their methylation in association with colon cancer. These included 147 hypermethylated and 27 hypomethylated genes. Of the hypermethylated genes, 10 were hypermethylated in 100% of colon cancers and many others were hypermethylated in more than 50% of colon cancers.
While epigenetic alterations are found in cancers, the epigenetic alterations in DNA repair genes, causing reduced expression of DNA repair proteins, may be of particular importance. Such alterations may occur early in progression to cancer and are a possible cause of the genetic instability characteristic of cancers.
Reduced expression of DNA repair genes disrupts DNA repair. This is shown in the figure at the 4th level from the top. (In the figure, red wording indicates the central role of DNA damage and defects in DNA repair in progression to cancer.) When DNA repair is deficient DNA damage remains in cells at a higher than usual level (5th level) and causes increased frequencies of mutation and/or epimutation (6th level). Mutation rates increase substantially in cells defective in DNA mismatch repair or in homologous recombinational repair (HRR). Chromosomal rearrangements and aneuploidy also increase in HRR defective cells.
Higher levels of DNA damage cause increased mutation (right side of figure) and increased epimutation. During repair of DNA double strand breaks, or repair of other DNA damage, incompletely cleared repair sites can cause epigenetic gene silencing.
Deficient expression of DNA repair proteins due to an inherited mutation can increase cancer risks. Individuals with an inherited impairment in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) have increased cancer risk, with some defects ensuring a 100% lifetime chance of cancer (e.g. p53 mutations). Germ line DNA repair mutations are noted on the figure’s left side. However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about 1 percent of cancers.
In sporadic cancers, deficiencies in DNA repair are occasionally caused by a mutation in a DNA repair gene but are much more frequently caused by epigenetic alterations that reduce or silence expression of DNA repair genes. This is indicated in the figure at the 3rd level. Many studies of heavy metal-induced carcinogenesis show that such heavy metals cause a reduction in expression of DNA repair enzymes, some through epigenetic mechanisms. DNA repair inhibition is proposed to be a predominant mechanism in heavy metal-induced carcinogenicity. In addition, frequent epigenetic alterations of the DNA sequences code for small RNAs called microRNAs (or miRNAs). miRNAs do not code for proteins, but can “target” protein-coding genes and
reduce their expression.
Cancers usually arise from an assemblage of mutations and epimutations that confer a selective advantage leading to clonal expansion (see Field defects in progression to cancer). Mutations, however, may not be as frequent in cancers as epigenetic alterations. An average cancer of the breast or colon can have about 60 to 70 protein-altering mutations, of which about three or four may be “driver” mutations and the remaining ones may be “passenger” mutations.
Metastasis
Metastasis is the spread of cancer to other locations in the body. The dispersed tumors are called metastatic tumors, while the original is called the primary tumor. Almost all cancers can metastasize. Most cancer deaths are due to cancer that has metastasized.
Metastasis is common in the late stages of cancer and it can occur via the blood or the lymphatic system or both. The typical steps in metastasis are local invasion, intravasation into the blood or lymph, circulation through the body, extravasation into the new tissue, proliferation and angiogenesis. Different types of cancers tend to metastasize to particular organs, but overall the most common places for metastases to occur are the lungs, liver, brain and the bones.
Metabolism
Normal cells typically generate only about 30% of energy from glycolysis, whereas most cancers rely on glycolysis for energy production (Warburg effect). But a minority of cancer types rely on oxidative phosphorylation as the primary energy source, including lymphoma, leukemia, and endometrial cancer. Even in these cases, however, the use of glycolysis as an energy source rarely exceeds 60%. A few cancers use glutamine as the major energy source, partly because it provides nitrogen required for nucleotide (DNA, RNA) synthesis. Cancer stem cells often use oxidative phosphorylation or glutamine as a primary energy source.